Introduction
The alignment system in ramen quantifies how similar two
or more microbial communities are from a functional perspective – not by
which species are present, but by which metabolite-to-metabolite
pathways they catalyse. align() supports three modes:
pairwise (two consortia), multiple
(all consortia in a set), and search (one query
consortium against every member of a set). All three return a
ConsortiumMetabolismAlignment (CMA) object containing
similarity scores and (depending on the mode) pathway correspondences, a
consensus network, or a ranked hit table.
This vignette covers the alignment system in depth. For a general
introduction to the package, see
vignette("ramen", package = "ramen").
Note on help lookup. If tibble or
pillar is loaded in your session, ?align may
resolve to pillar::align() first. To view the documentation
for this package’s generic, use ?ramen::align.
Mathematical formulation
This section gives the formal definitions of every quantity computed
by align(). Notation follows Hirt (2025), the master’s
thesis from which the package originated; equation numbers prefixed
(thesis eq. ...) refer to that document.
Notation
Let denote a collection of consortia (thesis eq. 1). For one consortium , write for its set of metabolites; for two consortia under comparison let with (thesis eq. 3). All assays are square matrices on this union metabolite space. We write and for the two binary assays under comparison; analogous symbols denote the Consumption, Production, and nSpecies assays. The norm is the sum of all entries; is the elementwise (Hadamard) product.
A pathway in ramen is a directed
metabolite-to-metabolite pair
such that some species in the consortium can consume
and some species (possibly the same one) can produce
.
Pathways are not biochemical reactions: they are
capabilities inferred from the Cartesian product of
each species’ uptake set and secretion set. A consortium of two species,
one consuming
and producing
,
contributes the four pathways
from only four distinct exchange events (two uptakes, two secretions).
See the Cross-product inflation caveat below.
Assay matrices
Each ConsortiumMetabolism stores eight assays (thesis
eq. 1, 2):
- Binary : if any species in realises pathway , else .
- nSpecies : number of distinct species supporting .
- Consumption : , the summed uptake flux of metabolite across all species participating in .
- Production : , analogously for secretion.
- EffectiveConsumption and EffectiveProduction : flux-corrected effective fluxes (see below).
- nEffectiveSpeciesConsumption and nEffectiveSpeciesProduction : Hill-1 effective number of contributing species (see below).
All eight are constructed as the Cartesian product for each species , then aggregated across species. This cross-product is the source of the density inflation discussed under Limitations and caveats.
Effective flux and effective species count
For a pathway with participating species , let be the fraction of total flux of type contributed by species . The Shannon entropy of this distribution is , and the Hill-1 perplexity is the effective number of species contributing to the pathway: unitless, , maximised when all species contribute equally.
ramen exposes both the perplexity itself and the
flux-corrected flux
(thesis eq. 2) as separate assays. They are two views of the same
network:
and analogously
for production. EffectiveConsumption and
EffectiveProduction carry the same units as
Consumption / Production (a flux,
larger when total flux is large and species contribute evenly);
nEffectiveSpeciesConsumption and
nEffectiveSpeciesProduction are unitless counts
mirroring nSpecies. The algebraic identity
holds cell-wise (modulo two-decimal
rounding); same for production. See the Hill-1 saturation
caveat for behaviour on small consortia.
Pairwise alignment metrics
Throughout, and are binary assays expanded to the union metabolite space, and are the Consumption, Production, and nSpecies assays for consortium .
FOS (Szymkiewicz-Simpson overlap)
Range
.
Asymmetric in size:
whenever the smaller binary network is a subset of the larger, which is
what motivates the coverage ratios below. Returns
by convention if either matrix is empty. This is the default
method.
Bray-Curtis (similarity, on combined fluxes)
This is equivalent to
Bray-Curtis on the stacked
abundance vector: summing the
differences of
and
separately and dividing by the combined
norms is algebraically the same as concatenating
and
into one vector and applying the standard Bray-Curtis formula. Range
,
with
at identical fluxes. The
clamp guards against floating-point near-negative zero; this is the
consequence of the B7 fix and is why brayCurtis = NA is
returned for unweighted CMs (the metric is undefined when fluxes are
unit placeholders). See the ?ramen::align
@note.
RedundancyOverlap (weighted Jaccard / Ruzicka)
Using and , Range . Collapse to Jaccard at . When all positive entries of and equal (every supported pathway is single-species) the numerator becomes the indicator intersection and the denominator becomes the indicator union , which is exactly . Small communities therefore yield identical Jaccard and RedundancyOverlap scores.
MAAS (Metabolic Alignment Aggregate Score)
A weighted convex combination of the four base metrics. Let be the set of metrics whose inputs are available (Bray-Curtis and RedundancyOverlap drop out for unweighted CMs). Default weights are . Disclosure: these defaults are not derived from data; they encode a soft preference for FOS as the primary metric and were not tuned by sensitivity analysis. A formal sensitivity sweep is deferred to the methods paper.
Permutation null model
For a metric
and observed value
,
a
-value
is computed by degree-preserving rewire of the query
binary network, treated as a directed graph
.
Let
denote the graph obtained from
by a sequence of edge swaps that preserve the in-degree and out-degree
sequence; igraph::keeping_degseq is used with
.
The permutation
-value
is
with
(default
).
The null hypothesis is “the observed similarity is no greater than
expected under random rewiring of
that preserves each metabolite’s participation count”.
Limitation (methods-paper concern, not a current correctness
issue). Degree-preserving rewire is the conventional null in
ecological network analysis but is not specifically motivated
for FBA-derived networks: edge presence in
reflects mass-balance constraints on the underlying genome-scale
metabolic models, plus the solver’s choice among alternate optima. A
null that respects those constraints (for example a configuration model
on the species-pathway bipartite graph, a species-set permutation, or a
randomised-abundance null in the MICOM sense) would be more defensible.
The current null is retained for reproducibility with prior
ramen versions; sensitivity to null-model choice is on the
methods-paper agenda.
When to use what: alignment vs functional groups
ramen exposes two superficially similar comparisons.
They answer different questions and are not
interchangeable.
Alignment (align(CM, CM),
align(CMS), align(CM, CMS)) compares two or
more named consortia by their metabolic exchange networks. The
unit of analysis is the consortium; the question is “how similar are
these communities by what they collectively do?”
Functional groups (functionalGroups())
cluster species by the Jaccard similarity of their per-species
pathway sets, either within a single consortium or pooled across a CMS.
The unit of analysis is the species; the question is “which species
play equivalent metabolic roles?” See
vignette("ramen", package = "ramen") for functional
groups.
In practice, run functionalGroups() first within a
consortium (or pooled across a CMS) to understand internal structure,
then align() to compare consortia.
Limitations and caveats
The metric formulas above are not a license to run alignment on arbitrary inputs. Several known issues affect interpretation.
Cross-product inflation. Pathways are inferred from the Cartesian product of each species’ uptake and secretion sets, not from biochemical reactions. For a consortium of two species and , the Binary assay has nonzero entries in a matrix, giving density from exchange events across distinct metabolites. This is a capability-level representation, not a mechanism-level one, and the inflation grows with per species. Compare consortia of similar size and composition; do not interpret raw Binary density as a mechanistic property.
Hill-1 saturation on small consortia.
nEffectiveSpeciesConsumption / nEffectiveSpeciesProduction store the
Hill-1 perplexity rounded to two decimals. For two-species consortia the
perplexity rarely exceeds
(one species typically dominates the flux at any given pathway), and the
round(., 2) step collapses near-degenerate distributions to
exactly
– indistinguishable from true single-species pathways. Because
EffectiveConsumption and EffectiveProduction are the elementwise product
,
the same saturation propagates to them: on small consortia they
degenerate toward the underlying Consumption / Production assays. Both
effective views are informative only on consortia where several species
contribute comparable flux to the same pathway.
Flux reversibility and no binarisation threshold.
ConsortiumMetabolism() admits any nonzero flux: a flux of
produces an edge identical to a flux of
.
FBA solvers (MiSoSoup, MICOM, cobrapy) routinely produce alternate
optima that differ only in sign of small fluxes near the noise floor;
under the current convention this turns a “consume
,
produce
”
pathway into “consume
,
produce
”,
which the Binary assay treats as a different pathway. No
flux_tolerance argument is currently exposed. For solver
outputs we recommend pre-filtering by solver tolerance (typically
for COBRA / cobrapy) before constructing CMs.
Bray-Curtis on unweighted CMs returns NA. The fix
for the floating-point near-negative-zero bug (B7) introduced an
explicit
clamp that propagates NA when fluxes are unit placeholders
rather than measured values. This is documented in the
align() @note. To get a non-NA
Bray-Curtis, build CMs from non-unit fluxes.
Alternative optima are not biological signal. MiSoSoup is a MILP enumerator: for a fixed metabolic model it returns multiple alternative optimal solutions to the same growth problem. Such alternatives typically share most pathways and produce very high pairwise FOS values that reflect solver consistency, not biological similarity. The same caveat applies to outputs from MICOM and cobrapy, where the choice of solver, tolerance, and tie-breaking affects which exchange fluxes appear in the solution. When interpreting overlap, compare consortia derived from different models, conditions, or experimental contexts; treat overlap between alternates of one model as a sanity check on the enumeration, not as ecology.
Minimum production flux drives the alignment.
Because pathways are inferred from the existence of any nonzero
production flux for a given species and metabolite, low-magnitude
secretions (whether biologically real or solver noise) determine the
topology of the Binary assay just as much as high-magnitude ones. This
is a design decision, not a bug: it preserves the set of
metabolic capabilities the consortium can realise. Users who
want to weight by magnitude should use Bray-Curtis or RedundancyOverlap
rather than FOS / Jaccard, and should consider thresholding fluxes
upstream of ConsortiumMetabolism().
Test data
We build six consortia from the bundled misosoup24
dataset to use throughout this vignette.
data("misosoup24")
cm_list <- lapply(seq_len(6), function(i) {
ConsortiumMetabolism(
misosoup24[[i]],
name = names(misosoup24)[i]
)
})
cms <- ConsortiumMetabolismSet(cm_list, name = "Demo")Pairwise alignment
Basic usage
align(CM, CM) compares two
ConsortiumMetabolism objects and returns a CMA with
Type = "pairwise".
cma <- align(cm_list[[1]], cm_list[[2]])
cma
#>
#> ── ConsortiumMetabolismAlignment
#> Name: "ac_A1R12_1 vs ac_A1R12_10"
#> Type: "pairwise"
#> Metric: "FOS"
#> Score: 0.7634
#> Query: "ac_A1R12_1", Reference: "ac_A1R12_10"
#> Coverage: query 0.763, reference 0.38
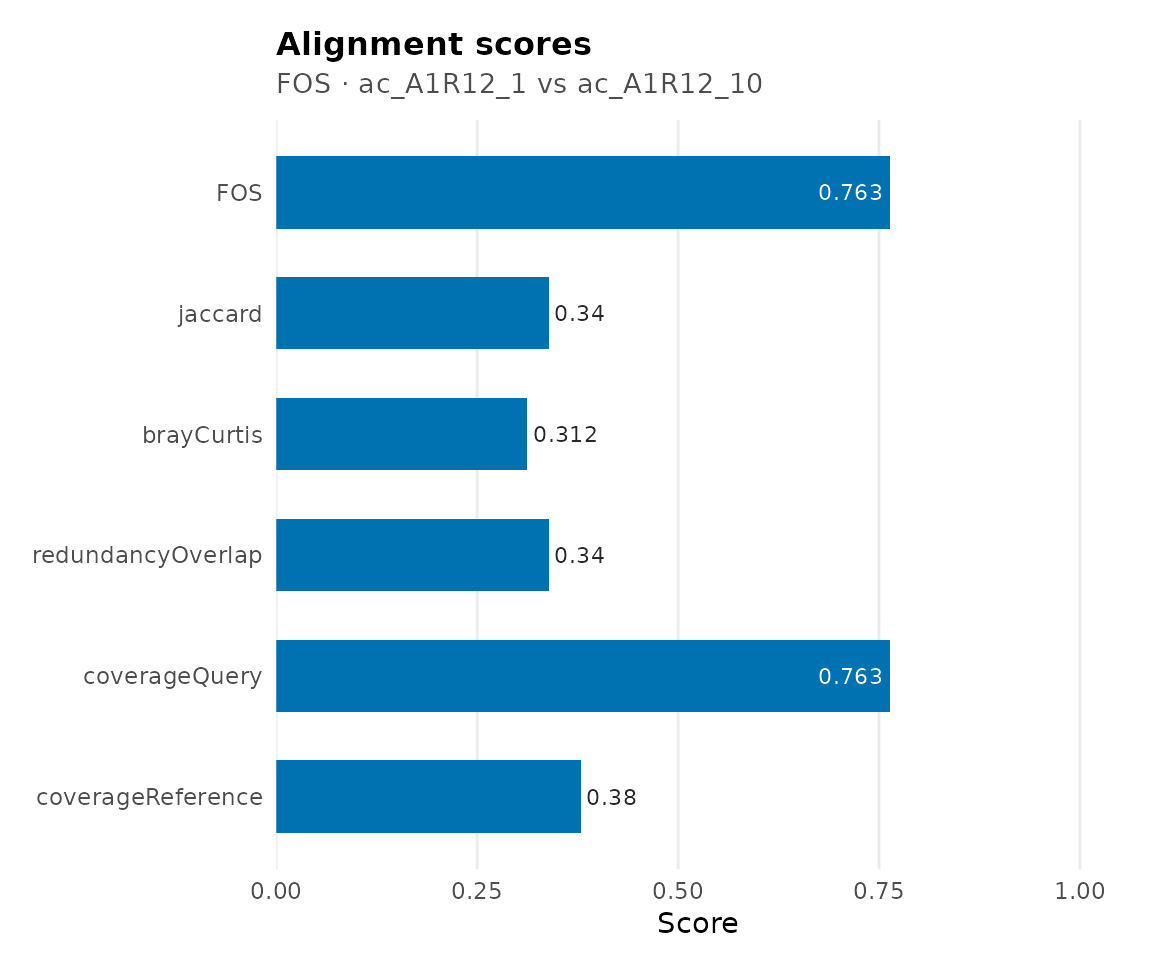
#> Pathways: 71 shared, 22 query-only, 116 reference-only.Similarity metrics
Five metrics are available via the method argument.
Regardless of which is selected as the primary score, all applicable
metrics are always computed and stored.
cma_fos <- align(cm_list[[1]], cm_list[[2]], method = "FOS")
cma_jac <- align(cm_list[[1]], cm_list[[2]], method = "jaccard")
scores(cma_fos)
#> $FOS
#> [1] 0.7634409
#>
#> $jaccard
#> [1] 0.3397129
#>
#> $brayCurtis
#> [1] 0.3115158
#>
#> $redundancyOverlap
#> [1] 0.3397129
#>
#> $coverageQuery
#> [1] 0.7634409
#>
#> $coverageReference
#> [1] 0.3796791| Metric | Description |
|---|---|
| FOS | Szymkiewicz-Simpson on binary matrices |
| Jaccard | Symmetric set similarity |
| Bray-Curtis | Flux-weighted similarity |
| Redundancy Overlap | Weighted Jaccard on nSpecies |
| MAAS | 0.4 FOS + 0.2 each of the rest |
The Metabolic Alignment Aggregate Score (MAAS) combines all four metrics. Weights are renormalised when some metrics are unavailable (e.g., unweighted networks lack Bray-Curtis):
cma_maas <- align(cm_list[[1]], cm_list[[2]], method = "MAAS")
scores(cma_maas)
#> $FOS
#> [1] 0.7634409
#>
#> $jaccard
#> [1] 0.3397129
#>
#> $brayCurtis
#> [1] 0.3115158
#>
#> $redundancyOverlap
#> [1] 0.3397129
#>
#> $coverageQuery
#> [1] 0.7634409
#>
#> $coverageReference
#> [1] 0.3796791
#>
#> $MAAS
#> [1] 0.5035647The FOS subset property
FOS uses the Szymkiewicz-Simpson coefficient, which divides by the smaller network. This means FOS = 1 whenever a small consortium is a strict functional subset of a larger one – even if the larger consortium has many additional pathways.
To detect this, ramen reports coverage
ratios alongside the similarity metrics:
scores(cma_fos)[c("FOS", "coverageQuery", "coverageReference")]
#> $FOS
#> [1] 0.7634409
#>
#> $coverageQuery
#> [1] 0.7634409
#>
#> $coverageReference
#> [1] 0.3796791-
coverageQuery: fraction of the query’s pathways found in the reference -
coverageReference: fraction of the reference’s pathways found in the query
When FOS is high but one coverage ratio is low, the alignment represents a subset relationship rather than true functional equivalence. For symmetric similarity, use Jaccard instead.
Pathway correspondences
The alignment classifies every metabolite-to-metabolite pathway as shared, unique to the query, or unique to the reference.
## All pathways in the alignment
head(pathways(cma))
#> # A tibble: 6 × 2
#> consumed produced
#> <chr> <chr>
#> 1 acald ac
#> 2 asp__L ac
#> 3 gly ac
#> 4 gthrd ac
#> 5 h2o2 ac
#> 6 h2s ac
## Shared pathways only
shared <- pathways(cma, type = "shared")
nrow(shared)
#> [1] 71
## Unique pathways (returns a list with $query and $reference)
unique_pw <- pathways(cma, type = "unique")
nrow(unique_pw$query)
#> [1] 22
nrow(unique_pw$reference)
#> [1] 116Permutation p-values
Statistical significance is assessed by degree-preserving network rewiring. The query network’s pathways are shuffled while preserving each metabolite’s degree, and the metric is recomputed under the null distribution. For MAAS, only the binary network topology is permuted; the weighted assays (Consumption, Production, nSpecies) remain fixed, so the null distribution reflects topological variation in the composite score.
cma_p <- align(
cm_list[[1]],
cm_list[[2]],
method = "FOS",
computePvalue = TRUE,
nPermutations = 99L
)
scores(cma_p)
#> $FOS
#> [1] 0.7634409
#>
#> $jaccard
#> [1] 0.3397129
#>
#> $brayCurtis
#> [1] 0.3115158
#>
#> $redundancyOverlap
#> [1] 0.3397129
#>
#> $coverageQuery
#> [1] 0.7634409
#>
#> $coverageReference
#> [1] 0.3796791
#>
#> $pvalue
#> [1] 0.01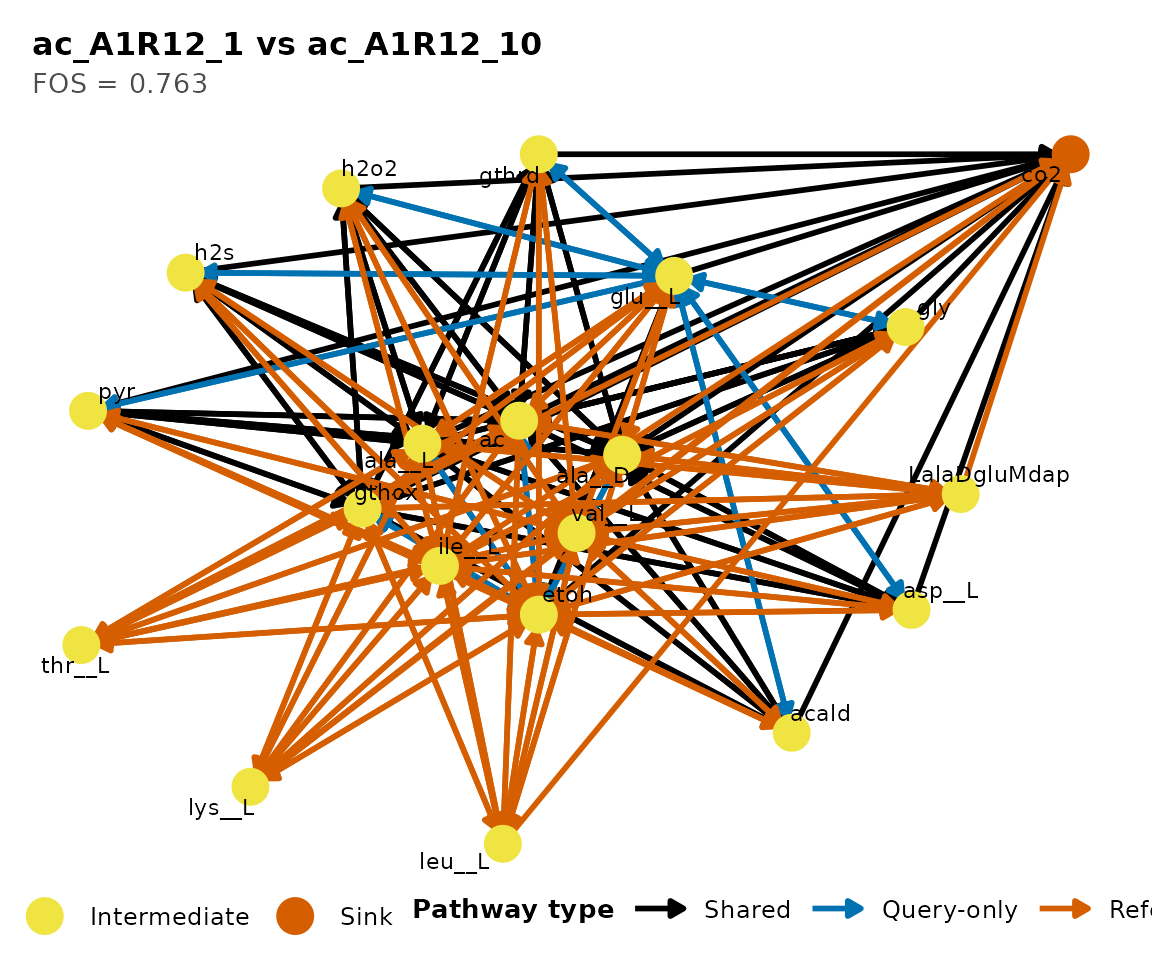
Multiple alignment
Aligning a consortium set
align(CMS) computes pairwise similarities across all
consortia in a ConsortiumMetabolismSet and returns a CMA
with Type = "multiple".
cma_mult <- align(cms)
cma_multSimilarity matrix
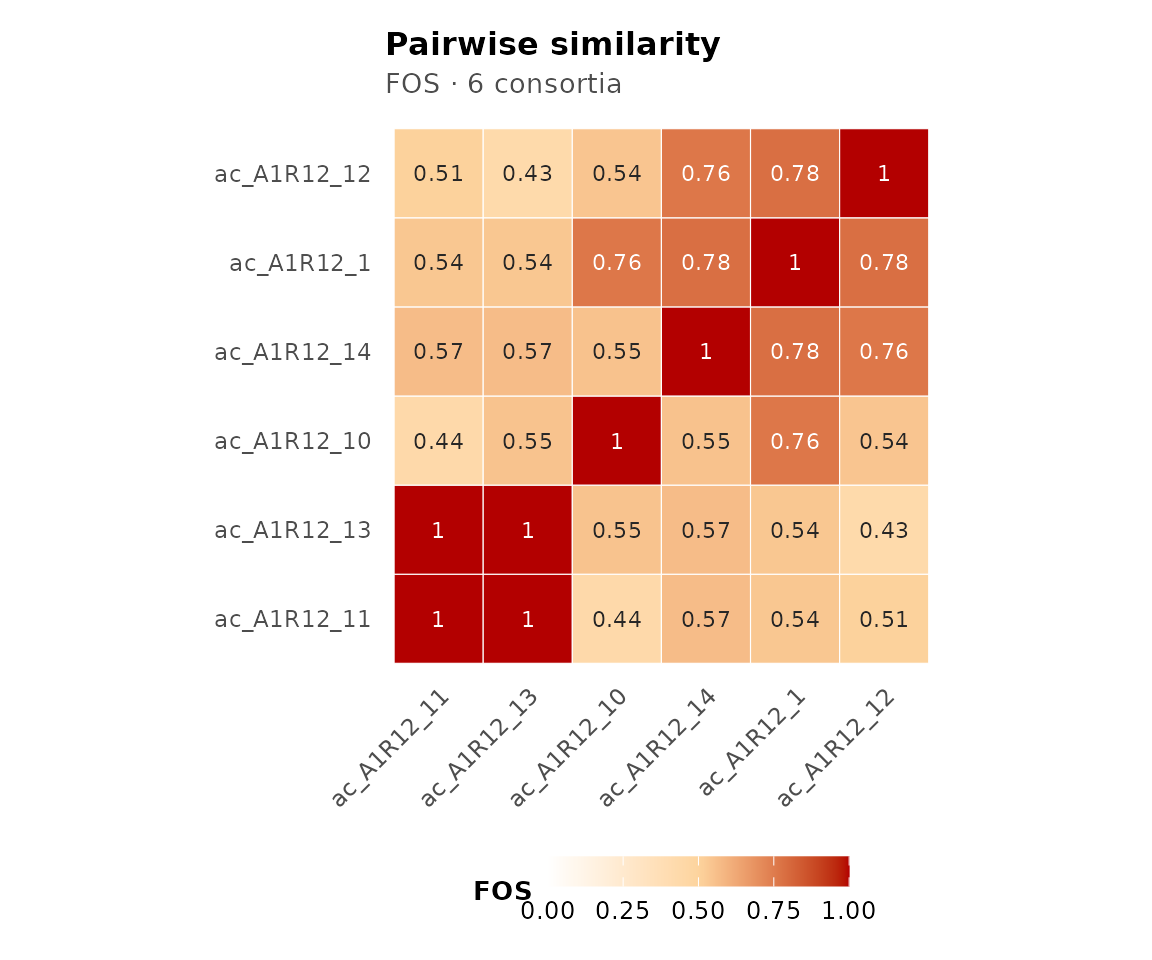
The similarity matrix is an n x n symmetric matrix with 1s on the diagonal. For FOS, this is derived from the pre-computed CMS overlap matrix:
round(similarityMatrix(cma_mult), 3)
#> ac_A1R12_1 ac_A1R12_10 ac_A1R12_11 ac_A1R12_12 ac_A1R12_13
#> ac_A1R12_1 1.000 0.763 0.538 0.785 0.538
#> ac_A1R12_10 0.763 1.000 0.438 0.542 0.549
#> ac_A1R12_11 0.538 0.438 1.000 0.507 1.000
#> ac_A1R12_12 0.785 0.542 0.507 1.000 0.430
#> ac_A1R12_13 0.538 0.549 1.000 0.430 1.000
#> ac_A1R12_14 0.785 0.551 0.567 0.764 0.567
#> ac_A1R12_14
#> ac_A1R12_1 0.785
#> ac_A1R12_10 0.551
#> ac_A1R12_11 0.567
#> ac_A1R12_12 0.764
#> ac_A1R12_13 0.567
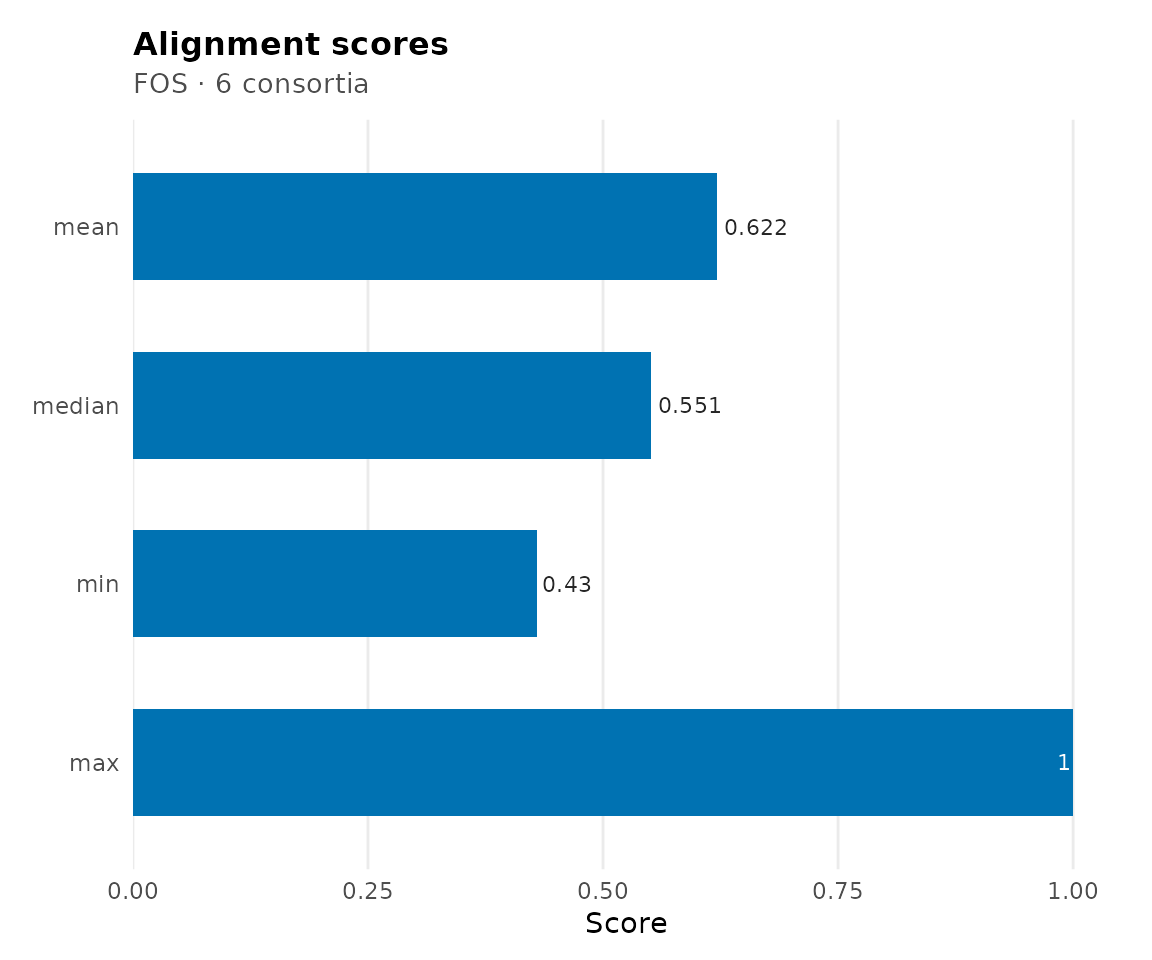
#> ac_A1R12_14 1.000Summary scores
For a multiple alignment, the primary score is the
median of all pairwise scores. scores()
returns full summary statistics:
scores(cma_mult)
#> $mean
#> [1] 0.6215862
#>
#> $median
#> [1] 0.5511811
#>
#> $min
#> [1] 0.4295775
#>
#> $max
#> [1] 1
#>
#> $sd
#> [1] 0.1597042
#>
#> $nPairs
#> [1] 15Consensus network and prevalence
Pathway prevalence counts how many consortia share each metabolite-to-metabolite pathway. This enables classification of pathways as core (present in most consortia) or niche (present in few).
prev <- prevalence(cma_mult)
head(prev[order(-prev$nConsortia), ])
#> consumed produced nConsortia proportion
#> 21 asp__L ac 6 1
#> 24 gly ac 6 1
#> 30 pyr ac 6 1
#> 56 asp__L ala__D 6 1
#> 59 gly ala__D 6 1
#> 65 pyr ala__D 6 1
## Distribution of prevalence
table(prev$nConsortia)
#>
#> 1 2 3 4 5 6
#> 100 75 55 51 14 30The pathways() method with
type = "consensus" returns the same information:
Database search
align(CM, CMS) compares a single query consortium
against every member of a set and returns a CMA with
Type = "search". This is the natural call for questions
like “which consortium in my database is most functionally similar to
this query?”
Basic search
We hold out the first consortium as a query and search it against a database built from the remaining five:
query <- cm_list[[1]]
db <- ConsortiumMetabolismSet(cm_list[-1], name = "db")
hits <- align(query, db)
#> Searching 5 consortia using "FOS".
hits
#>
#> ── ConsortiumMetabolismAlignment
#> Name: "ac_A1R12_1 vs CMS (5 consortia)"
#> Type: "search"
#> Metric: "FOS"
#> Score: 0.7849
#> Query: "ac_A1R12_1", Top hit: "ac_A1R12_12" (of 5 consortia)The top-hit name and score sit on ReferenceName /
PrimaryScore. The full ranked table is stored in
Scores$ranking:
ranking <- scores(hits)$ranking
head(ranking)
#> # A tibble: 5 × 8
#> reference score FOS jaccard brayCurtis redundancyOverlap coverageQuery
#> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
#> 1 ac_A1R12_12 0.785 0.785 0.451 0.635 0.451 0.785
#> 2 ac_A1R12_14 0.785 0.785 0.497 0.553 0.497 0.785
#> 3 ac_A1R12_10 0.763 0.763 0.340 0.312 0.340 0.763
#> 4 ac_A1R12_11 0.538 0.538 0.226 0.500 0.226 0.538
#> 5 ac_A1R12_13 0.538 0.538 0.270 0.515 0.270 0.538
#> # ℹ 1 more variable: coverageReference <dbl>Each row holds the reference consortium’s name, the primary
score (under the requested method), all four
individual metric columns, and the coverage ratios against the query.
Rows are pre-sorted by score in descending order.
Top-K hits
Use topK to truncate the ranked table – useful when the
database is large and only the best matches matter:
hits_top3 <- align(query, db, topK = 3L)
#> Searching 5 consortia using "FOS".
scores(hits_top3)$ranking
#> # A tibble: 3 × 8
#> reference score FOS jaccard brayCurtis redundancyOverlap coverageQuery
#> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
#> 1 ac_A1R12_12 0.785 0.785 0.451 0.635 0.451 0.785
#> 2 ac_A1R12_14 0.785 0.785 0.497 0.553 0.497 0.785
#> 3 ac_A1R12_10 0.763 0.763 0.340 0.312 0.340 0.763
#> # ℹ 1 more variable: coverageReference <dbl>similarityMatrix() returns a 1 x n row vector (or 1 x
topK if truncated), with the query name as the row label:
round(similarityMatrix(hits_top3), 3)
#> ac_A1R12_12 ac_A1R12_14 ac_A1R12_10
#> ac_A1R12_1 0.785 0.785 0.763Pathway correspondences always reflect the single overall top hit,
regardless of topK:
Choosing metrics
By default all four base metrics are computed for every database
member. For large databases, restricting metrics skips the
weighted-assay expansion and can be substantially faster:
hits_fos <- align(query, db, metrics = "FOS")
#> Searching 5 consortia using "FOS".
scores(hits_fos)$ranking[,
c("reference", "score", "brayCurtis")
]
#> # A tibble: 5 × 3
#> reference score brayCurtis
#> <chr> <dbl> <dbl>
#> 1 ac_A1R12_12 0.785 NA
#> 2 ac_A1R12_14 0.785 NA
#> 3 ac_A1R12_10 0.763 NA
#> 4 ac_A1R12_11 0.538 NA
#> 5 ac_A1R12_13 0.538 NAColumns for skipped metrics remain in the ranking table but are
filled with NA, making the schema stable across calls.
Significance of the top hit
As in pairwise alignment, computePvalue = TRUE runs a
degree-preserving permutation test. For database search, the test is
applied only to the top hit – a BLAST-style convention that keeps the
cost comparable to a single pairwise p-value:
hits_p <- align(
query,
db,
computePvalue = TRUE,
nPermutations = 99L
)
#> Searching 5 consortia using "FOS".
hits_p
#>
#> ── ConsortiumMetabolismAlignment
#> Name: "ac_A1R12_1 vs CMS (5 consortia)"
#> Type: "search"
#> Metric: "FOS"
#> Score: 0.7849
#> P-value: "0.01"
#> Query: "ac_A1R12_1", Top hit: "ac_A1R12_12" (of 5 consortia)The show() output reports the top hit, its score, and
the p-value directly; scores(hits_p) gives the full numeric
breakdown including pvalue.
If statistical confidence is needed for several top hits, re-run
align() in pairwise mode against each candidate
individually.
Accessor reference
| Accessor | Alignment type | Returns |
|---|---|---|
scores() |
all | Named list of scores (+ $ranking for search) |
pathways() |
all | data.frame of all pathways |
pathways(type = "shared") |
pairwise, search | shared pathways |
pathways(type = "unique") |
pairwise, search | list(query, reference) |
pathways(type = "consensus") |
multiple | data.frame with prevalence |
similarityMatrix() |
multiple, search | n x n or 1 x n numeric matrix |
prevalence() |
multiple | data.frame with nConsortia |
metabolites() |
all | Character vector of metabolites |
Type guards prevent misuse – for example, calling
pathways(type = "consensus") on a pairwise alignment raises
an informative error.
Session info
sessionInfo()
#> R version 4.6.0 (2026-04-24)
#> Platform: x86_64-pc-linux-gnu
#> Running under: Ubuntu 24.04.4 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
#>
#> locale:
#> [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
#> [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
#> [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
#> [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: UTC
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] ramen_0.99.0 BiocStyle_2.40.0
#>
#> loaded via a namespace (and not attached):
#> [1] tidyselect_1.2.1 viridisLite_0.4.3
#> [3] dplyr_1.2.1 farver_2.1.2
#> [5] viridis_0.6.5 Biostrings_2.80.0
#> [7] S7_0.2.2 ggraph_2.2.2
#> [9] fastmap_1.2.0 SingleCellExperiment_1.34.0
#> [11] lazyeval_0.2.3 tweenr_2.0.3
#> [13] digest_0.6.39 lifecycle_1.0.5
#> [15] tidytree_0.4.7 magrittr_2.0.5
#> [17] compiler_4.6.0 rlang_1.2.0
#> [19] sass_0.4.10 tools_4.6.0
#> [21] utf8_1.2.6 igraph_2.3.1
#> [23] yaml_2.3.12 knitr_1.51
#> [25] labeling_0.4.3 graphlayouts_1.2.3
#> [27] S4Arrays_1.12.0 DelayedArray_0.38.1
#> [29] RColorBrewer_1.1-3 TreeSummarizedExperiment_2.20.0
#> [31] abind_1.4-8 BiocParallel_1.46.0
#> [33] withr_3.0.2 purrr_1.2.2
#> [35] BiocGenerics_0.58.0 desc_1.4.3
#> [37] grid_4.6.0 polyclip_1.10-7
#> [39] stats4_4.6.0 ggplot2_4.0.3
#> [41] scales_1.4.0 MASS_7.3-65
#> [43] SummarizedExperiment_1.42.0 cli_3.6.6
#> [45] rmarkdown_2.31 crayon_1.5.3
#> [47] ragg_1.5.2 treeio_1.36.1
#> [49] generics_0.1.4 ape_5.8-1
#> [51] cachem_1.1.0 ggforce_0.5.0
#> [53] parallel_4.6.0 BiocManager_1.30.27
#> [55] XVector_0.52.0 matrixStats_1.5.0
#> [57] vctrs_0.7.3 yulab.utils_0.2.4
#> [59] Matrix_1.7-5 jsonlite_2.0.0
#> [61] bookdown_0.46 IRanges_2.46.0
#> [63] S4Vectors_0.50.0 ggrepel_0.9.8
#> [65] systemfonts_1.3.2 dendextend_1.19.1
#> [67] tidyr_1.3.2 jquerylib_0.1.4
#> [69] glue_1.8.1 pkgdown_2.2.0
#> [71] codetools_0.2-20 gtable_0.3.6
#> [73] GenomicRanges_1.64.0 tibble_3.3.1
#> [75] pillar_1.11.1 rappdirs_0.3.4
#> [77] htmltools_0.5.9 Seqinfo_1.2.0
#> [79] R6_2.6.1 textshaping_1.0.5
#> [81] tidygraph_1.3.1 evaluate_1.0.5
#> [83] lattice_0.22-9 Biobase_2.72.0
#> [85] memoise_2.0.1 bslib_0.10.0
#> [87] Rcpp_1.1.1-1.1 gridExtra_2.3
#> [89] SparseArray_1.12.2 nlme_3.1-169
#> [91] xfun_0.57 fs_2.1.0
#> [93] MatrixGenerics_1.24.0 pkgconfig_2.0.3